

Structure modeling and molecular dynamics simulation in protein-protein interaction study
- Faculty of Biology – Biotechnology, University of Science, VNU-HCM
- Vietnam National University Ho Chi Minh City
Abstract
Protein is a vital macromolecule that contributes to the living system. Functionally, proteins not only work independently, but also form a close network via protein-protein interactions (PPIs), the decisive factor in most biological processes in living organisms. Nevertheless, the study of PPIs has encountered many obstacles due to the lack of experimental structures of the complexes or proteins involved in the interactions. Thanks to the support of bioinformatics tools, methods, and algorithms, the prediction of protein interaction structure has emerged as a potential solution to solve the above difficulties. Molecular docking and molecular dynamics simulation (MDs) are two commonly used methods for predicting PPIs because of their ability to model structure and approximate biological processes, which are consistent with the natural state of the realistic system. Applying these bioinformatics advances has shortened the time, effort, and research costs for scientists. This review provides information on the protein-protein complex structure prediction method and some tools to assist in previous studies. In addition, the force field component of the molecular kinetics simulation method is highlighted, and simulation programs are extensively used.
MỞ ĐẦU
Protein là đại phân tử sinh học quan trọng, đóng vai trò kiến tạo nên hệ thống sống. Các báo cáo chỉ ra rằng hơn 80% protein không hoạt động đơn lẻ mà thường tương tác với các phân tử khác như DNA, RNA hoặc protein để thực hiện các chức năng chuyên biệt trong tế bào 1. Thuật ngữ “tương tác protein” mô tả sự tiếp xúc vật lý giữa protein và đối tác tương tác, đồng thời mô tả các tương tác của chúng khi thực hiện chức năng 2. Việc xác định các tương tác giữa protein-protein (protein-protein interactions – PPIs) là vô cùng quan trọng trong việc đào sâu vào những cơ chế của sinh học tế bào, khám phá và thiết kế các phương pháp trị liệu mới, kỹ thuật protein và nghiên cứu đột biến gen3. Một số phương pháp thực nghiệm hiệu năng cao như hệ thống lai đôi ở nấm men (Yeast two-hybrid – Y2H), đồng tủa miễn dịch (co-immunoprecipitation – co-IP), phương pháp khối phổi xác định phức hợp protein (Mass spectrometric protein complex identification – MS-PCI) đã và đang được sử dụng để phát hiện tương tác giữa các protein 4, 5, 6. Các phương pháp này đóng góp nhiều vào bộ dữ liệu PPI ở nhiều loài nhưng lại tốn kém, mất nhiều thời gian và công sức. Ngoài ra, sự ảnh hưởng bởi yếu tố ngoại cảnh của điều kiện thí nghiệm cũng làm tăng tỷ lệ dương tính và âm tính giả 7. Từ đó, bằng cách kết hợp những thông tin có được từ thực nghiệm và sự hỗ trợ của sinh học tính toán, các kết quả tương tác protein-protein hay tương tác phối tử-thụ thể ngày càng có sự chính xác và chất lượng tốt hơn 8.
Cấu trúc protein đơn lẻ hay phức hợp protein-protein đã đem lại nhiều thông tin và cung cấp kiến thức về quá trình hoạt động của phân tử. Tuy vậy, trong thực tế, protein luôn chuyển động và chức năng sinh học cũng như tương tác protein-phối tử phụ thuộc hoàn toàn vào sự linh động của các phân tử thành phần. Để theo dõi được động học của protein, các phương pháp thực nghiệm đã được áp dụng và nắm bắt được chuyển động của protein ở mức từ femto giây tới nano giây như tinh thể hóa tia X độ phân giải siêu cao (ultra resolution X-ray crystallography), cộng hưởng từ hạt nhân siêu nhanh (ultra fast NMR), kính hiển vi lực nguyên tử (atomic force microscopy – AFM) hay phổ huỳnh quang (fluorescence spectroscopy). Tương tự với quá trình xác định tương tác protein, các phương pháp nêu trên cũng gây khó khăn cho các nhà nghiên cứu vì đòi hỏi nhiều thiết bị tiên tiến hay kĩ thuật có độ khó cao để theo dõi được động học của protein9. Mô phỏng động lực học phân tử (Molecular dynamic simulations – MDs) dự đoán chuyển động của các nguyên tử trong protein theo thời gian dựa trên mô hình vật lý mô tả các lực tác động lên chúng10. Các mô phỏng này có thể đưa cho chúng ta những góc nhìn mới về các quá trình sinh học quan trọng như thay đổi cấu trạng, tương tác phối tử hay gấp cuộn protein. Vì thế, MDs đang ngày càng được sử dụng rộng rãi kết hợp cùng với các phương pháp thực nghiệm nhằm quan sát được chuyển động với thời gian dài và chính xác hơn.
Bài tổng quan này được thực hiện với mục tiêu giới thiệu về các phương pháp dự đoán cấu trúc tương tác protein-protein in silico, đặc biệt là phương pháp mô phỏng động lực học phân tử, tập trung vào khía cạnh trường lực, các gói chương mô phỏng đang được sử dụng rộng rãi và ứng dụng trong nghiên cứu phát triển thuốc.
MÔ HÌNH HÓA CẤU TRÚC TƯƠNG TÁC PROTEIN-PROTEIN
Tương tác protein-protein giữ chức năng quan trọng trong hầu hết các hoạt động sinh học của tế bào, bao gồm dẫn truyền tín hiệu, sự di chuyển của tế bào, nhận diện kháng nguyên và tiêu diệt các tác nhân xâm nhiễm. Tuy đã có những cải tiến về sinh học phân tử cũng như hệ gen học (genomics), chức năng của phần lớn protein vẫn chưa được biết rõ11. Theo Jansen, sự tương tác giữa những protein đã biết hay chưa biết chức năng có thể đóng góp hiệu quả vào việc giải mã những bí ẩn này12. Hiện nay, hai phương pháp được phát triển và sử dụng cho việc xây dựng mô hình cấu trúc tương tác protein-protein là docking phân tử (molecular docking), hay mô hình hóa không khuôn mẫu (template-free modeling – TFM), và mô hình hóa dựa trên khuôn mẫu (template-based modeling – TBM) (Figure 1).
Mô hình hóa cấu trúc tương tác không khuôn mẫu hay Docking phân tử
Trong mô hình hóa cấu trúc không khuôn mẫu (template-free modeling – TFM), hay docking phân tử (molecular docking), phức hợp cấu trúc tương tác được xây dựng bằng cách lắp ráp các cấu trúc đã biết của các phần tử tương tác, được tìm thấy hoặc dự đoán ở dạng protein đơn, thông qua cách tìm kiếm hệ thống và lựa chọn các hướng gắn khác nhau. Phương pháp này dựa trên sự phù hợp về đặc tính lí hoá và tính bổ sung hình học ở bề mặt của phức hợp13. Bề mặt phân tử có thể được mô tả theo diện tích bề mặt có thể tiếp cận với dung môi và mô tả theo bề mặt tiếp xúc với phân tử mục tiêu của nó. Tính bổ sung giữa hai bề mặt phân tử được đánh giá dựa trên sự phù hợp về hình dạng, hỗ trợ bằng việc xác định các rãnh hoặc hốc bổ sung trên bề mặt phân tử của những mục tiêu cho quá trình docking 14, 15, 16. Cách tiếp cận này hiệu quả với phức hợp có bề mặt bổ sung nhau một cách rõ ràng với diện tích bề mặt lớn (>1400 Å) và có ưu thế về tính kỵ nước17. Phương pháp này mô phỏng tốt phần lớn các cấu trúc phức hợp; tuy nhiên, sự hạn chế trong các thuật toán lấy mẫu (sampling) không gian có thể tạo ra những kết quả dương tính giả có tính bổ sung bề mặt tốt nhưng lại khác xa với cấu trúc tự nhiên và phụ thuộc vào độ chính xác của hàm chấm điểm 13, 14, 15, 16, 17, 18. Các phương pháp tìm kiếm cho docking protein-protein có thể được phân thành ba hướng tiếp cận bao gồm: tìm kiếm toàn diện tổng thể (exhaustive global search), tương thích hình dạng vị trí cục bộ (local shape feature matching) và tìm kiếm ngẫu nhiên (randomized search). Trong khi đó, các thuật toán lấy mẫu tiên tiến hơn so với docking phân tử nhỏ là mô hình hóa bằng mạng ClustENM kết hợp HADDOCK, lấy mẫu Monte Carlo nâng cao, hay phương pháp Aether đang ngày càng được sử dụng rộng rãi19, 20, 21, 22. ClusPro và HADDOCK là hai trong số những sever docking thường được sử dụng.
- ClusPro (https://cluspro.bu.edu/login.php?redir=/home.ph) dự đoán phức hợp protein-protein dựa trên phương pháp docking thân cứng (rigid docking) 23. Quá trình docking gồm ba bước bao gồm: (1) Phối tử được docking cứng với thụ thể trong 70000 bước xoay; (2) Trong 70000 bước xoay đó, chọn ra 1000 tổ hợp tịnh tiến/xoay có điểm thấp nhất; (3) Một nghìn tổ hợp đó được phân cụm với bán kính RMSD < 9 Å, nghĩa là vị trí phối tử có nhiều lân cận (neighbor) trong bán kính 9 Å được chọn và trở thành trung tâm cụm (centroid). ClusPro cho ra kết quả được xếp hạng theo điểm tổng số năng lượng thấp nhất được tính dựa trên hàm năng lượng có sẵn và độ lớn của cụm cấu trạng; trong đó, cấu trúc trung tâm được chọn làm đại diện cho từng cụm cấu trạng. ClusPro cho phép người dùng tùy chỉnh giới hạn tìm kiếm để trả kết quả theo số lượng mong muốn24.
- HADDOCK (High ambiguity driven protein-protein docking) (https://www.bonvinlab.org/software/) là công cụ docking linh hoạt (flexible docking) nhiều loại đại phân tử từ protein đến glycan 25. HADDOCK hoạt động theo quy trình: (1) Docking thân cứng các tiểu phần một cách ngẫu nhiên vào các không gian với khoảng cách tương đối và tối thiểu năng lượng để tạo phức hợp; (2) Docking bán linh hoạt và tinh chỉnh các cấu trúc có số điểm cao; (3) Docking linh hoạt thực hiện việc mô phỏng động lực học phân tử các mô hình từ bước trước. Cách xây dựng mô hình của HADDOCK là tích hợp tổng thể các thông tin sinh hóa, tin sinh học và thông tin cấu trúc liên quan26. HADDOCK là một trong những công cụ docking đầu tiên áp dụng đến giới hạn tương tác không rõ ràng (ambiguous interaction restraints – AIR). Thông qua các thông tin thực nghiệm có sẵn như dịch chuyển hóa học NMR, đột biến, HDX, các amino acid tham gia vào tương tác sẽ được chọn và từ đó có thể xây dựng phức hợp tương tác một cách chính xác hơn.
Mô hình hóa cấu trúc tương tác dựa trên khuôn mẫu
Trong phương pháp hình hóa dựa trên khuôn mẫu (template-based modeling – TBM), phức hợp cấu trúc tương tác được xây dựng bằng dựa trên cấu trúc phức hợp khuôn mẫu đã được xác định từ cơ sở dữ liệu. Phương pháp này đã được dùng để dự đoán cấu trúc bậc bốn của protein chuỗi đơn dựa trên nguyên tắc sự tương đồng trình tự có thể dẫn đến tương đồng về cấu trúc 27. Các bước thực hiện tổng quát như sau: (1) Tìm kiếm một hoặc nhiều khuôn mẫu phù hợp; (2) Sắp gióng cột trình tự mục tiêu với trình tự khuôn mẫu; (3) Xây dựng mô hình ban đầu cho cấu trúc truy vấn bằng cách giữ nguyên các phân đoạn cấu trúc từ các vùng sắp gióng cột của khuôn mẫu; (4) Thay thế các chuỗi bên phù hợp với trình tự truy vấn; (5) Xây dựng các vùng loop và vùng kết thúc chuỗi; (6) Tinh chỉnh mô hình để thu được toàn bộ cấu trúc. Dù TBM đã thể hiện được một số tiến bộ so với TFM, phương pháp này vẫn tồn tại những hạn chế nổi bật như độ chính xác của mô hình sẽ giảm nếu độ tương đồng của trình tự mục tiêu và trình tự khuôn mẫu thấp hơn 40% cũng như thiếu phương pháp tinh chỉnh phức hợp protein28, 29, 30. Hai trong những công cụ được sử dụng để dự đoán PPI theo phương pháp TBM là SWISS-MODEL và AlphaFold2.
- SWISS-MODEL (https://swissmodel.expasy.org) là công cụ mô phỏng tự động tiên phong trong quá trình xây dựng mô hình 3D của protein từ năm 1993 cho đến nay31. Cấu trúc phức hợp protein-protein thông qua thuật toán máy học (machine learning) có giám sát support vector Machines (SVM), kết hợp bởi những thông tin về vùng tương tác bảo tồn, gom cụm cấu trúc, và những thông số khác để xây dựng giá trị chất lượng cấu trúc bậc bốn (auaternary structure quality estimate – QSQE)32. Những phức hợp có mức độ tương đồng cao thường sở hữu cấu trúc bậc bốn và phương thức tương tác như nhau; qua đó, Bertoni và cộng sự đã đề xuất dấu chỉ PPI (PPI fingerprint), một hàm số thể hiện được sự bảo tồn bề mặt tương tác qua quá trình tiến hóa. Phân tích dấu chỉ PPI giúp cung cấp thêm các thông tin quan trọng cho dự đoán cấu hình bậc bốn của protein.
- AlphaFold2 bên cạnh dự đoán cấu trúc protein đơn lẻ còn có thể giúp chúng ta xây dựng mô hình tương tác dựa trên các phức hợp có sẵn. Công cụ hoạt động theo cách sử dụng các trình tự có mối liên hệ tiến hóa, sắp gióng cột nhiều trình tự; sau đó sử dụng Evoformer để tìm mối liên hệ giữa trình tự và cấu trúc khuôn mẫu33. Evoformer xem protein như các đám mây đơn phân di chuyển xung quanh mạng lưới để tạo ra mô hình 3D; cuối cùng các tinh chỉnh cục bộ được thực hiện để đưa ra kết quả 34.
Khuyết điểm lớn nhất của SWISS-MODEL hay AlphaFold2 là cả hai đều bị ảnh hưởng bởi thông tin về các phức hợp protein-protein có sẵn để dự đoán chính xác. Kết quả của SWISS-MODEL hoàn toàn được mô phỏng dựa trên cấu trúc khuôn mà người dùng chọn; vì thế, việc lựa chọn được cấu trúc khuôn mẫu đáng tin cậy cũng như kiến thức về tương tác là vô cùng cần thiết. Trong khi đó, AlphaFold2 mặc dù đã cải tiến hơn với việc chỉ dựa vào trình tự và dự đoán tương tác thông qua thông tin di truyền nhưng vẫn còn hạn chế trong việc đưa ra cấu trúc protein-protein chính xác. Việc này chủ yếu đến từ bộ dữ liệu để huấn luyện cho AlphaFold-Multimer vẫn còn ít và chưa bao quát được toàn bộ các trường hợp có thể xảy ra. Tuy nhiên, các nhà nghiên cứu vẫn đang tiếp tục phát triển, cải thiện AlphaFold và đã đạt được một số kết quả hứa hẹn35, 36, 37, 38.

Hai phương pháp dự đoán cấu trúc phức hợp. (A) Mô hình hóa không dựa trên khuôn mẫu (TFM); (B) Mô hính hóa dựa trên khuôn mẫu (TBM).
MÔ PHỎNG ĐỘNG LỰC HỌC PHÂN TỬ
Mặc dù được xem như là một công cụ vô cùng hữu dụng trong những nghiên cứu về mối quan hệ giữa cấu trúc và chức năng, docking phân tử vẫn tồn tại nhiều nhược điểm. Điều này được bắt nguồn từ việc docking phân tử chỉ được xem như một phương pháp lấy mẫu cứng, trong khi protein lại vô cùng linh động, và không hề phản ánh được sự thay đổi của phân tử khi bị ảnh hưởng bởi phân tử khác (ion, dung môi,…) hay môi trường (pH, điện trường,…) 39. Một số tương tác phân tử không thể được dự đoán chính xác, bao gồm tác động của dung môi hay thay đổi entropy 40, 41. Từ đó, docking phân tử cần được kết hợp với một số kĩ thuật khác để có thể cung cấp những kết quả đáng tin cậy hơn, và một trong số đó chính là mô phỏng động lực học phân tử (Molecular Dynamics Simulations – MDs). Từ những năm 50 của thế kỷ trước, các nhà khoa học đã thể hiện mối quan tâm đến cách thức hoạt động ở cấp độ nguyên tử liên quan đến chức năng của một phân tử 42. Kể từ đó mô phỏng động học phân tử được xem như một giải pháp hữu hiệu để nghiên cứu sự chuyển động không ngừng của các nguyên tử nhờ vào sự hỗ trợ của máy tính. Năm 1977, McCammon và cộng sự đã thực hiện nghiên cứu đầu tiên để có cái nhìn sâu sắc về động học của protein ức chế trypsin tuyến tụy bò, nghiên cứu này đã làm thay đổi quan điểm xem protein như một hệ cứng nhắc 43, 44. Figure 2 thể hiện số lượng các công bố về sinh học cấu trúc có sử dụng mô phỏng động học kể từ năm 1977 cho thấy tính hữu ích của mô phỏng động học phân tử trong nghiên cứu, đặc biệt trong lĩnh vực khoa học thần kinh khi được dùng để nghiên cứu các protein quan trọng với tín hiệu thần kinh 45.

Số lượng bài báo có sử dụng mô phỏng động học phân tử trong giai đoạn từ 1977 đến 2017
Nguyên lý và nhiệm vụ của mô phỏng động học phân tử
Mô phỏng động học là kỹ thuật nghiên cứu sự chuyển động của một hệ gồm rất nhiều nguyên tử, được xem như các hạt, trong một khoảng thời gian ngắn bằng việc sử dụng phương trình chuyển động của Newton. Trong khoảng thời gian mô phỏng, sự biến đổi của các hạt trong hệ được tính toán bằng tích phân phương trình chuyển động của chúng:
Với m là khối lượng, a là gia tốc, F là lực tác dụng lên hạt thứ i.
Điểm mạnh của mô phỏng động học phân tử là cho phép hệ ở trạng thái động, điều này phù hợp với thực tế là các đại phân tử sinh học nói chung hay protein nói riêng tồn tại ở trạng thái động, cung cấp phương tiện để mô hình hóa tính linh hoạt và những thay đổi về hình dạng của protein trong các quá trình sinh học ở các điều kiện khác nhau của môi trường44.
Trường lực
Trường lực được định nghĩa là một biểu thức toán học mô tả sự phụ thuộc năng lượng của một hệ thống vào tọa độ của từng nguyên tử cấu thành hệ. Có nhiều cách phân loại và dạng trường lực sẵn có hiện nay 47, 48, 49, tuy nhiên đều sử dụng cùng một biểu thức điển hình cho trường lực:
Biểu thức trên, nhìn chung, được tách biệt thành hai dạng là năng lượng nội phân tử và liên phân tử. Năng lượng liên kết bao gồm ba phần tử đầu tiên trong biểu thức trên là E được tính bằng tổng độ dài tất cả các liên kết giữa hai nguyên tử và E tính toán độ rộng các góc liên kết. Cách khai triển hai phần tử này tương tự như cách tính độ co giãn của lò xo trong định luật Hooke50. Năng lượng xoay góc nhị diện ϕ và ψ (E) tương ứng với n góc nhị diện có mặt trong chuỗi 48. Trong khi đó, năng lượng liên phân tử, đóng vai trò quan trọng trong tương tác protein-protein, đề cập đến các tương tác tĩnh điện và Van der Waals (VdW) giữa những nguyên tử không tạo liên kết trực tiếp. Năng lượng từ tương tác VdW được tính dựa trên thế năng Lennard-Jones 12-6, mô hình lực đẩy điện tử và các tương tác phân tán 51. Cuối cùng, định luật Coulomb được sử dụng để mô tả tương tác tĩnh điện giữa một cặp nguyên tử đại diện bởi đại lượng E51.
Hiện nay có ba mô hình phân tử chính đã được phát triển gồm: trường lực toàn nguyên tử 52, 53, trường lực thô hóa 54, và trường lực kết hợp 55, 56. Các nghiên cứu về động học phân tử đang sử dụng một cách phổ biến bốn họ trường lực dành cho protein là: AMBER, CHARMM, GROMOS, OPLS-AA47. Những trường lực này khác nhau về dữ liệu và quy trình sử dụng để tham số hóa. Tuy nhiên, Jorgensen và Tirado đã khẳng định các trường lực hiện có vẫn cung cấp các kết quả đáng tin cậy57. Table 1 tóm tắt một số trường lực hiện có trong các họ trường lực nêu trên.
Các trường lực hiện có trong mô phỏng động lực học đại phân tử sinh học
|
Họ trường lực |
Trường lực |
Mô tả |
|
CHARMM |
CHARMM22 |
Một trường lực toàn nguyên tử dành cho protein |
|
CHARMM27 |
Trường lực dành cho nucleic acid và lipid | |
|
AMBER |
ff19SB |
Đang được sử dụng rộng rãi để mô phỏng protein |
|
GLYCAM |
Mô phỏng carbohydrate | |
|
ff14SBonlysc |
Là trường lực tốt nhất đối với mô phỏng trong dung môi ẩn | |
|
GROMOS |
GROMOS53a6 |
Trường lực dùng trong mô phỏng carbohydrate |
|
GROMOS45a3 |
Sử dụng cho các tập hợp lipid như màng tế bào, micelle | |
|
GROMOS45a4 |
Cải thiện cách tham số hóa nucleic acid | |
|
OPLS-AA |
OPLS3 |
Mô phỏng động học phân tử nhỏ và protein |
Các chương trình mô phỏng động lực học phân tử
- CHARMM (Chemistry at HARvard Molecular Mechanic, https://www.charmm.org/) được phát triển từ 40 năm trước, căn bản tập trung vào các đại phân tử sinh học như protein, nucleic acid, lipid, carbohydrate v.v.. Chương trình cung cấp một lượng lớn bộ công cụ tính toán bao gồm nhiều phương pháp lấy mẫu cấu hình và ước tính năng lượng tự do, tối thiểu năng lượng, động học và kỹ thuật phân tích cũng như khả năng xây dựng mô hình 63, 64. CHARMM được nhà phát triển xây dựng với ba yếu tố giúp làm nên độ hiệu quả của gói mô phỏng này là (1) hoàn tất công việc trong khoảng thời gian ngắn nhất có thể, (2) dữ liệu đầu vào được đơn giản hóa tối đa, (3) nhà phát triển quan tâm đến việc thiết kế giao diện chương trình dễ hiểu và dễ chỉnh sửa64.
- AMBER (Assisted Model Building with Energy Refinement, https://ambermd.org/) là chương trình mô phỏng giúp phân tích hiệu quả động học của các phân tử như protein, nucleic acid và carbohydrate. AMBER không phải là một chương trình đơn lẻ mà là một tập hợp các gói chương trình được thiết kế để hoạt động cùng nhau. Quy trình thực hiện gồm ba bước chính: chuẩn bị hệ thống, mô phỏng và phân tích kết quả. Figure 3 tóm tắt quy trình thông tin trong gói mô phỏng AMBER. Quy trình này được phân tích chi tiết trong bài tổng quan “Chương trình mô phỏng sinh học phân tử Amber” của David và cộng sự và tài liệu hướng dẫn sử dụng Amber2265. AMBER hỗ trợ một trường lực tổng quát cho các phân tử hữu cơ nhỏ, được dùng để phân tích phức hợp của phân tử nhỏ với protein hoặc nucleic acid66.

Quy trình thông tin đầu vào trong gói mô phỏng AMBER. pdb4amber: chương trình giúp chuyển đổi file ở định dạng khác sang định dạng pdb để tương thích với LeaP; prepareforleap: một tùy chọn trong công cụ cpptraj có cùng chức năng như pdb4amber, thích hợp cho carbohydrate; Antechamber: chương trình giúp xây dựng trường lực cho các phân tử hữu cơ nhỏ như thuốc, amino acid được biến đổi; pyMSMT: gói mô hình hóa vị trí gắn kim loại của hệ hỗn hợp; mdgx: chương trình tạo các thông số trường lực liên kết; LeaP: chương trình tạo ra hệ mô phỏng trong Amber; sander: chương trình tối thiểu hóa năng lượng và động lực học phân tử; pmemd: một phiên bản của sander được tối ưu hóa; MMPBSA.py: code Python để phân tích năng lượng từ quá trình mô phỏng động lực học phân tử; FEW: chương trình tính toán năng lượng tự do của hệ protein-phối tử; mdout_analyze.py: file đầu ra của chương trình sander và pmemd; cpptraj: chương trình phân tích quỹ đạo và dữ liệu.
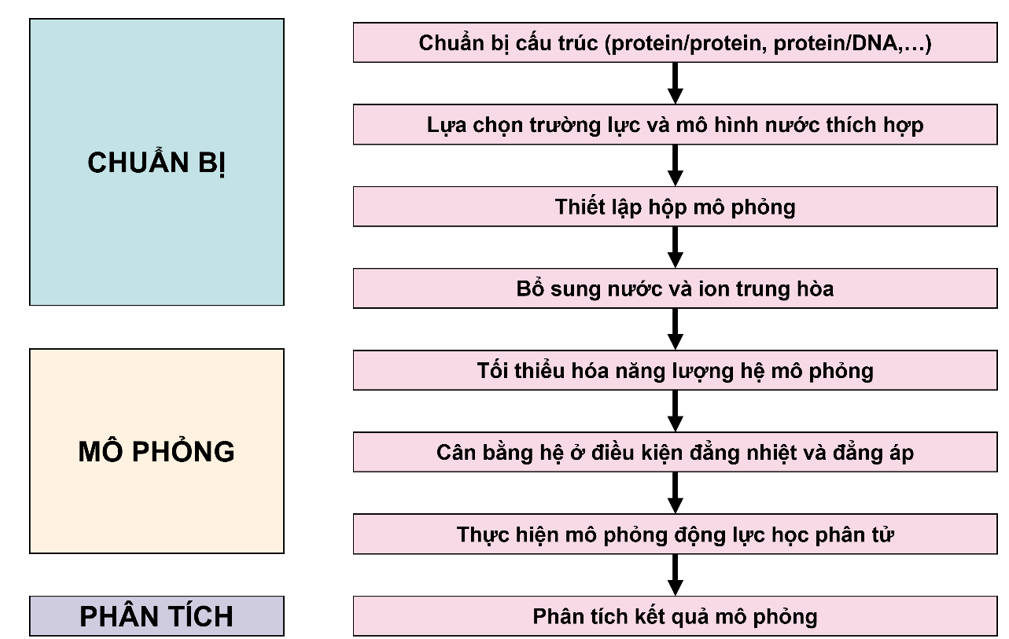
- GROMACS (GROningen MAchine for Chemical Simulations, https://www.gromacs.org/) là một hệ thống mã nguồn mở được sử dụng rộng rãi nhất trong mô phỏng động học phân tử sinh học như protein, lipid, nucleic acid, cũng như các phân tử polymer phi sinh học67. GROMACS được đánh giá là một phần mềm nhanh, linh hoạt, miễn phí và thân thiện với người dùng68, 69. Hiệu suất của chương trình mô phỏng được nâng cao hơn kể từ phiên bản GROMACS 2019 khi có sự hỗ trợ của phần mềm tích hợp gmx_api. Phần mềm này cung cấp một giao diện Python cho phép kết nối các câu lệnh của GROMACS với các công cụ phân tích của bên thứ ba70. Chi tiết các bước chạy mô phỏng trên GROMACS được thể hiện ở Figure 4 và bài tổng quan của Smith và cộng sự71.

Các bước chạy mô phỏng bằng GROMACS. Phân tử mục tiêu được chuẩn bị cho GROMACS bằng cách áp dụng các thông tin về trường lực và mô hình dung môi (thường là nước). Sau đó, hộp mô phỏng được thiết lập và bổ sung thêm dung môi, ion trung hòa toàn bộ điện tích hệ. Hệ mô phỏng này sau đó được tối ưu hóa năng lượng cũng như trải qua các bước cân bằng hệ ở những điều kiện đẳng nhiệt (NVT) và đẳng áp (NPT) trước khi thực hiện bước mô phỏng cuối.
- NAMD (Nanoscale Molecular Dynamics, http://www.ks.uiuc.edu/Research/ namd/) là gói chương trình được thiết kế để mô phỏng hiệu suất cao của các hệ thống phân tử sinh học lớn. Một mục tiêu quan trọng của NAMD là cung cấp cho người dùng một giao diện thống nhất trên tất cả các nền tảng, từ laptop đến máy tính để bàn và cả trên hệ thống siêu máy tính. Điều này được thực hiện dựa trên hệ thống lập trình song song Charm72. NAMD sử dụng thuật toán sao chép nhiều lần (Multiple Copy Algorithms – MCA) hỗ trợ tốt hơn trong việc lấy mẫu cấu hình, tính toán năng lượng tự do hay tinh chỉnh các trạng thái chuyển tiếp 73. Ngoài ra, một ưu điểm của NAMD là giao diện tập lệnh Tcl cung cấp tính linh hoạt tối đa cho người dùng có thể sửa đổi hoặc triển khai MCA một cách độc lập ngay cả khi đang chạy mô phỏng 72, 73.
ỨNG DỤNG TRONG NGHIÊN CỨU PHÁT TRIỂN THUỐC
Các bất thường xảy ra trong tương tác protein-protein có thể dẫn đến các loại bệnh khác nhau, trong đó có ung thư74. Nghiên cứu tương tác protein-protein bằng công cụ tin sinh học đã hỗ trợ mạnh mẽ trong công cuộc phát triển thuốc chữa bệnh 75, 76. Chiến lược phòng ngừa và điều trị bằng chất ức chế cộng hóa trị nổi lên như một phương pháp tiềm năng vì ít khả năng thúc đẩy quá trình kháng thuốc 77. Năm 2019, Khuchtumur và cộng sự phát triển chất ức chế TEAD-347 nhắm vào tương tác giữa protein Yap và TEAD thông qua tương tác với một cysteine bảo tồn trong túi palmitate78. Ngoài ra, mạng lưới tương tác protein-protein được xây dựng nhằm nghiên cứu các con đường truyền tín hiệu và dự đoán chức năng của protein chưa biết79.
Hiện nay mô phỏng động lực học phân tử được dùng rộng rãi trong nghiên cứu protein như sự tương tác protein-protein hay các con đường truyền tín hiệu80. Ví dụ, cặp thụ thể protein G (G protein-coupled receptor – GPCR) là loại thụ thể quan trọng với các loại thuốc, hơn một phần tư số thuốc nhắm mục tiêu vào loại thụ thể này80, 81, 82. Năm 2020, 24 loại thuốc mới nhắm mục tiêu GPCR đã được phê duyệt lâm sàng và 44 loại thuốc mới đang trong giai đoạn thử nghiệm83. Mô phỏng động lực học phân tử cung cấp thông tin chi tiết về ba cơ chế của GPCR bao gồm: sự thay đổi cấu hình trong trạng thái hoạt động và không hoạt động, tương tác của GPCR với phối tử/chất ức chế, tác động của lipid đối với cấu trúc của GPCR84. Một ví dụ khác là trong nghiên cứu cách thức các thể đột biến của SARS-CoV-2 tương tác với thụ thể ACE2. Cụ thể, bốn biến thể chứa các đột biến đã được nghiên cứu, trong đó đột biến E484K và N501Y làm tăng ái lực liên kết, khiến khả năng lây nhiễm cao hơn85. Song song đó, Acharya và cộng sự đã thực hiện mô phỏng động học trên 8000 hợp chất nhằm sàng lọc các chất ức chế mạnh protein S và xác định được 77 hợp chất thuốc phân tử nhỏ tiềm năng trong điều trị COVID-1986.
KẾT LUẬN
Mối quan tâm về cấu trúc phức hợp protein-protein được xem như vấn đề nổi trội, giúp các nhà nghiên cứu có cái nhìn sâu hơn vào cơ chế tương tác trong các quá trình sinh học. Do các khó khăn trong việc giải cấu trúc thực nghiên nên việc dự đoán cấu trúc phức hợp protein-protein bằng công cụ tin sinh học đã trở thành một phương pháp thay thế tối ưu. Tuy nhiên, trong thực tế các phân tử sinh học tham gia vào quá trình sinh học trong cơ thể luôn ở trạng thái động. Vì vậy, phương pháp mô phỏng động lực học phân tử đã được sử dụng từ những năm 70 của thế kỷ trước để hỗ trợ cho nghiên cứu sự tương tác liên quan đến chức năng của protein. Từ đó việc kết hợp phương pháp mô hình hóa cấu trúc phức hợp protein-protein và mô phỏng động lực học phân tử đã trở thành chìa khóa để mở rộng các nghiên cứu mang tính ứng dụng về sau.
DANH MỤC TỪ VIẾT TẮT
PPIs: protein-protein interactions
MDs: molecular dynamics simulation
Y2H: yeast two-hybrid
co-IP: co-immunoprecipitation
MS-PCI: mass spectrometric protein complex
AFM: atomic force microscopy
TFM: template-free modeling
TBM: template-based modeling
VdW: Van der Waals
CHARMM: chemistry at Harvard molecular mechanic
AMBER: assisted model building with energy refinement
GROMACS: groningen machine for chemical simulations
NAMD: nanoscale molecular dynamics
MCA: multiple copy algorithms
GPCR: G protein-coupled receptor
XUNG ĐỘT LỢI ÍCH
Các tác giả cam kết không có xung đột lợi ích.
ĐÓNG GÓP CỦA CÁC TÁC GIẢ
Nguyễn Văn Minh Thường: viết, tổng hợp và chỉnh sửa bản thảo
Lý Cẩm Tú: viết, tổng hợp và chỉnh sửa bản thảo
Đinh Thuận Thiên: lên ý tưởng, viết, tổng hợp và chỉnh sửa bản thảo
Trần Văn Hiếu: lên ý tưởng, tham gia chỉnh sửa bản thảo và chấp thuận bản thảo
Tất cả các tác giả đồng ý với bản thảo cuối cùng.