

Protein structure modeling using cloud-based servers
- Laboratory of Biosensors
- Department of Molecular and Environmental Biotechnology, Faculty of Biology and Biotechnology, University of Science, Vietnam National University – Ho Chi Minh City, Vietnam
Abstract
The structure of a protein plays an important role in determining its function. Deciphering how proteins fold has thus been a puzzle for nearly a half-century. Structure determination methods such as X-ray crystallography, nuclear magnetic resonance spectroscopy or electron microscopy are tedious and time-consuming; meanwhile, new-generation sequencing methods are producing massive amounts of protein data in a matter of days. To bridge the protein sequence-structure gap, methods and algorithms for predicting protein 3D structure starting from only its amino acid sequence have been developed. In recent years, machine learning has been of interest to the research community thanks to its work in analyzing intrinsic features of proteins to predict their structure, novel computational methods capable of modeling protein structures to near experimental accuracy. Accurately predicted structures are used for drug and antibody design, understanding protein-protein interactions, and with other molecules. This review provides information about multiple prediction methods and tools for protein structure prediction.
GIỚI THIỆU
Cấu trúc protein đầu tiên được xác định là cấu trúc myoglobin với phương pháp tinh thể hóa tia X. Từ đó đến nay đã có nhiều phương pháp khác hỗ trợ xác định cấu trúc protein như chụp cộng hưởng từ hạt nhân (NMR), kính hiển vi điện tử (EM); tuy nhiên, các phương pháp này rất phức tạp, tốn nhiều thời gian và sức lao động; cụ thể đến từ việc tinh thể hóa cấu trúc protein1, 2. Các protein có cấu trúc bất định (structural disorder), tồn tại xoắn xuyên màng, hay có nhiều vùng uốn và motif coil-coil trong cấu trúc rất khó để tinh thể hóa3. Ngược lại, công nghệ giải trình tự thông lượng cao (high-throughput sequencing) ngày càng phát triển 4; điều này đã tạo nên sự chênh lệch số lượng trình tự-cấu trúc trên cơ sở dữ liệu. Nhằm rút ngắn khoảng cách cấu trúc-trình tự, các phương pháp dự đoán cấu trúc protein đang được quan tâm.
Bài tổng quan này được thực hiện với mong muốn đưa ra cái nhìn tổng quát về những phương pháp dự đoán cấu trúc protein như mô phỏng tương đồng (homology modeling), xâu chuỗi/nhận diện kiểu gấp cuộn (threading/fold recognition), dự đoán ab initio, giới thiệu một số công cụ đã và đang được phát triển để phục vụ quá trình dự đoán cấu trúc protein (Figure 1).
DỰ ĐOÁN CẤU TRÚC PROTEIN DỰA TRÊN KHUÔN (TEMPLATE-BASED METHOD – TBM)

Quy trình dự đoán cấu trúc protein
Mô phỏng tương đồng (Homology modeling)
Phương pháp mô phỏng tương đồng được sử dụng lần đầu bởi Browne dựa trên nguyên tắc những protein có độ tương đồng cao hơn 30% thì sẽ có cấu trúc tương tự nhau5. Mô hình hóa tương đồng thường được ưu tiên khi cần dự đoán cấu trúc cho một protein, với quá trình dự đoán các bước chính như sau:
-
Tìm kiếm và lựa chọn trình tự tham chiếu : Trình tự tham chiếu có thể được tìm bằng cách sử dụng các công cụ như BLAST hoặc PSI-BLAST 6 để tìm các trình tự tương đồng trên PDB. Một số tiêu chí để lựa chọn trình tự tham chiếu là độ tương đồng cao, cùng họ protein, có quan hệ tiến hóa gần. Với độ tương đồng thấp hơn 30% thì kết quả sắp gióng cột tự động có thể không tối ưu do các đột biến điểm, đột biến thêm đoạn hoặc mất đoạn 7, 8. -
Mô hình hóa khung sườn : Có nhiều phương án tiếp cận để mô hình hóa cấu trúc khung sườn bao gồm tổ hợp khung cứng (rigid body assembly), chia thành phân đoạn ngắn theo tọa độ Cα, tích hợp các thông tin ràng buộc không gian về độ dài và góc liên kết (spatial restraint) và đột biến cấu trúc tham chiếu 9. -
Mô hình hóa đoạn uốn (loop) : Mô hình đoạn uốn có thể được mô phỏng dựa trên cấu trúc có sẵn hoặc dự đoánab initio theo phương pháp Monte Carlo hoặc mô phỏng động lực học phân tử 10. -
Mô hình hóa chuỗi bên : Các chuỗi bên thường được dự đoán theo phương pháp trích xuất tham chiếu từ thư viện rotamer (rotamer library) 11. Thư viện này được xây dựng từ các cấu trúc protein có sẵn với độ phân giải cao, với chuỗi bên phù hợp với cấu trúc mục tiêu được chọn dựa theo hàm năng lượng.
Với phương pháp mô phỏng tương đồng, chất lượng mô hình bị ảnh hưởng bởi độ tương đồng trình tự. Khi độ tương đồng giảm xuống khoảng 30-40% thì quá trình sắp gióng cột khó khăn hơn, dẫn đến hệ quả là 80% nguyên tử khung sườn có độ lệch RMSD khoảng 3.5 Å12.
Hiện nay, một số công cụ cho phép dự đoán mô hình hóa tương đồng có thể kể đến như là SWISS-MODEL và MODELLER.
SWISS-MODEL (https://swissmodel.expasy.org/) là một server dự đoán cấu trúc protein dựa trên phương pháp mô phỏng tương đồng bao gồm ba chế độ dự đoán: tự động, sắp gióng cột, và dự án 13. Công cụ QMEAN cũng được tích hợp để đánh giá chất lượng mô hình dự đoán14.
MODELLER (https://salilab.org/modeller/) là một phần mềm độc lập (stand-alone program) tương thích với nhiều hệ điều hành. MODELLER cho phép tích hợp các thông tin cưỡng chế không gian (spatial restraints) vào quá trình mô hình hóa cấu trúc và kết quả dự đoán được đánh giá bằng thuật toán DOPE15.
SWISS-MODEL và MODELLER đều là những công cụ được sử dụng nhiều trong dự đoán cấu trúc protein. Tuy nhiên, SWISS-MODEL lại đem đến cho người dùng giao diện thân thiện và tự động hơn so với giao diện câu lệnh của MODELLER. Mặc dù vậy, MODELLER lại để người dùng theo dõi và điều khiển từng bước trong quá trình mô phỏng cấu trúc.
Phương pháp xâu chuỗi/Nhận diện kiểu gấp cuộn (Threading/Fold Recognition)
Phương pháp xâu chuỗi (threading) ra đời nhằm hỗ trợ dự đoán các protein có độ tương đồng thấp hơn 30%, dựa trên thực tế rằng các trình tự khác biệt hoàn toàn có thể có cấu trúc giống nhau16. Phương pháp này còn được biết đến như là phương pháp “so sánh trình tự với cấu trúc”.
Các bước chính của phương pháp xâu chuỗi bao gồm:
-
Xây dựng thư viện cấu trúc tham chiếu : Các cấu trúc và kiểu gấp cuộn protein có thể được thu nhận từ các cơ sở dữ liệu như PDB, CATH, FSSP, và SCOP17. -
Thiết kế hàm chấm điểm : Hàm chấm điểm được thiết kế tốt sẽ bao gồm các tiêu chí về đột biến, tiềm năng tương tác giữa các cặp amino acid, mức độ phù hợp của trình tự với cấu trúc bậc hai, các điểm phạt do mất đoạn/thêm đoạn,… 18 -
Sắp gióng xâu chuỗi (threading alignment) : Sắp gióng trình tự mục tiêu với các kiểu gấp cuộn tiềm năng được đánh giá bởi hàm chấm điểm. -
Xây dựng mô hình 3D : Cấu trúc được mô hình hóa bắt đầu từ việc chọn các kiểu sắp gióng xâu chuỗi có tiềm năng lớn nhất rồi mô hình hóa dựa theo cấu trúc tham chiếu đã chọn.
Tuy vậy, phương pháp threading vẫn tồn tại nhược điểm chủ yếu nằm ở bản chất cấu trúc protein, cụ thể là tính thoái hóa kiểu cấp cuộn và những thiếu sót về dữ liệu thông tin môi trường khi thiết kế hàm chấm điểm19.
Một số công cụ cho phép dự đoán cấu trúc protein bằng phương pháp xâu chuỗi/nhận diện gấp cuộn có thể kể đến bao gồm I-TASSER và MUSTER.
I-TASSER (https://zhanggroup.org/I-TASSER/) là server dự đoán cấu trúc được phát triển bởi Yang Zhang Lab dựa trên thuật toán LOMETS để tìm kiếm cấu trúc tham chiếu, Monte Carlo để mô phỏng và phân nhóm cấu trạng bằng SPICKER. Các cấu trúc có ΔG thấp tiếp tục được cải thiện chất lượng mô phỏng động lực học phân tử với FG-MD và ModRefiner 20.
MUSTER (https://zhanggroup.org/MUSTER/) được xây dựng dựa trên thuật toán xâu chuỗi MUSTER (MUlti-Sources ThreadER) nhận diện tham chiếu từ PDB bằng quy hoạch động. Trong đó, các thông tin về trình tự và cấu trúc được tích hợp vào quá trình tìm kiếm bao gồm: hồ sơ trình tự, cấu trúc bậc hai, hồ sơ cấu trúc phụ thuộc độ sâu, xác suất tiếp xúc dung môi, góc nhị diện khung sườn, và ma trận điểm kị nước 21.
Cả hai công cụ trên đều được phát triển bởi nhóm nghiên cứu Yang Zhang. So với MUSTER dự đoán cấu trúc protein dựa trên việc xâu chuỗi tìm kiếm trình tự và mô phỏng bằng MODELLER, I-TASSER sử dụng thuật toán mô phỏng động lực học tích hợp với khả năng dự đoán tâm hoạt tính và bản thể học của gene (gene ontology).
DỰ ĐOÁN CẤU TRÚC PROTEIN KHÔNG DỰA TRÊN KHUÔN (TEMPLATE-FREE METHOD – TFM)
Thực tế cho thấy một số lượng lớn dữ liệu trình tự không hề tương đồng với những họ protein đã biết trước đó. Vì thế, phương pháp dự đoán không dựa trên khuôn mẫu đã ra đời (ab initio). Một thuật toán dự đoán ab initio tốt phải thỏa mãn 3 tiêu chí sau: tích hợp hàm năng lượng có độ chính xác cao; phương pháp lấy mẫu hiệu quả; và tiêu chí lựa chọn kết quả tốt nhất từ những cấu trúc dự đoán tiềm năng 22.
Hàm năng lượng
Hàm năng lượng biểu diễn cấu trúc protein dưới dạng các giá trị toán học, từ đó dẫn hướng cho thuật toán dự đoán tìm ra các cấu hình có ΔG thấp nhất. Hàm năng lượng có thể được phân thành 2 nhóm:
-
Hàm năng lượng dựa trên vật lý học : Dự đoán cấu trúc protein được thực hiện bằng cách kết hợp hàm năng lượng vật lý cơ học lượng tử hay trường lực với các phương pháp lấy mẫu cấu hình nhanh23, 24, 25. Một số trường lực cơ lý thuyết bao gồm AMBER, CHARMM, OPLS, GROMOS26. -
Hàm năng lượng thiết kế từ kiến thức và các cấu trúc phân mảnh có sẵn : Các hàm này được thiết kế bằng cách khai thác các đặc tính năng lượng không phụ thuộc trình tự và không thuộc trình tự, thế tương tác phụ thuộc khoảng cách nguyên tử và xu hướng hình thành cấu trúc bậc hai từ các cấu trúc sẵn có trên PDB27.
Phương pháp lấy mẫu (tìm kiếm) cấu hình
-
Mô phỏng Monte Carlo (MC) : Mô phỏng MC cổ điển yêu cầu rất nhiều tài nguyên máy tính và dễ gặp sai sót do các trạng thái ổn định giả (meta-stable state). Một số phương pháp cải tiến để khắc phục hạn chế trên gồm MC trao đổi lặp (REM) và MC tối thiểu hóa (MCM)28, 29. -
Mô phỏng động lực học phân tử (MDs) : MDs phản ánh tương đối chính xác quá trình gấp cuộn của protein nhưng lại tốn nhiều tài nguyên và thời gian nên thường xuyên được sử dụng để nghiên cứu sự gấp cuộn của protein30. Mô phỏng động lực động lực học phân tử tăng tốc (accelerated molecular dynamics – aMD) là phương pháp lấy mẫu cải thiện từ MD cổ điển bằng cách giảm thiểu rào chắn năng lượng, phân cách các trạng thái khác nhau của hệ thống, giảm thời gian mô phỏng 31.
Phương pháp chọn mô hình kết quả
Có hai nhóm phương án tiếp cận chính để lựa chọn mô hình dự đoán bao gồm phương án dựa trên năng lượng (energy-based method) và phương án dựa trên năng lượng tự do (free-energy based method). Các phương pháp dựa trên năng lượng bao gồm:
-
Chấm điểm dựa trên hàm năng lượng vật lý : Do năng lượng của cấu hình tự nhiên thấp hơn các cấu trúc mồi (structure decoy), các hàm năng lượng vật lý được sử dụng để hỗ trợ lựa chọn mô hình kết quả 32. -
Chấm điểm bằng hàm năng lượng dựa trên kiến thức : Hàm chấm điểm dựa trên kiến thức có thể là hàm năng lượng thô hóa (coarse-grained), trong đó các amino acid được biểu diễn dưới dạng một hoặc một vài nguyên tử đại diện. Cấu trúc dự đoán không thể hoàn toàn giống cấu trúc tự nhiên; vì thế yêu cầu về hàm chấm điểm cho phép phát hiện những cấu hình gần với tự nhiên (near-native) được đặt ra, tuy nhiên việc phát triển các hàm này vẫn còn gặp nhiều khó khăn33. -
Chấm điểm dựa trên độ tương hợp giữa trình tự và cấu trúc : Phương pháp này không phụ thuộc vào năng lượng mà dựa vào độ tương hợp giữa trình tự với cấu trúc. Một trong các cách tiếp cận đầu tiên là ứng dụng điểm xâu chuỗi 3D-1D của Luthy dựa trên việc gán cho từng amino acid các điểm số được tính bởi các thông số môi trường, bao gồm: diện tích vùng kị nước, tỷ lệ chuỗi bên bao phủ bởi nguyên tử O và N, và cấu trúc bậc hai lân cận34. Ngoài ra, hàm sai số bình phương của Colovos để mô tả các tương tác không cộng hóa trị giữa các cặp nguyên tử CC, CN, CO, NN, NO và OO cũng được sử dụng 35. Nhiều phương pháp và chương trình mới đã ra đời để cải thiện phương pháp của Luthy như VERIFY3D và GenTHREADER 22. -
Phân nhóm các cấu trúc trung gian : Các nhóm cấu trúc được phân cụm dựa trên ΔG có kích thước lớn thường giống với cấu trúc tự nhiên nhất 36. Các thuật toán phân cụm phổ biến hiện nay là SPICKER và ROSETTA 37.
Một số công cụ cho phép dự đoán cấu trúc protein bằng phương pháp ab initio có thể kể đến bao gồm Phyre2, RoseTTAFold, MetaFold và nổi tiếng nhất hiện nay là AlphaFold2.
Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2) kết hợp nhiều thuật toán dự đoán với nhau, chẳng hạn như HHblits, mô hình hidden Markov và HHsearch để dự đoán cấu trúc protein dựa trên khuôn38. Đối với những vùng không có cấu trúc tham chiếu, thuật toán poing được sử dụng để mô hình hóa khung sườn của protein; poing tăng tốc quá trình gấp cuộn bằng cách sử dụng động lực học Langevin và mô hình dung môi đặc biệt để nhanh chóng đẩy các amino acid kị nước vào trong phần lõi; cuối cùng, các chuỗi bên amino acid sẽ được dựng lên trên phần khung sườn đã được dự đoán39.
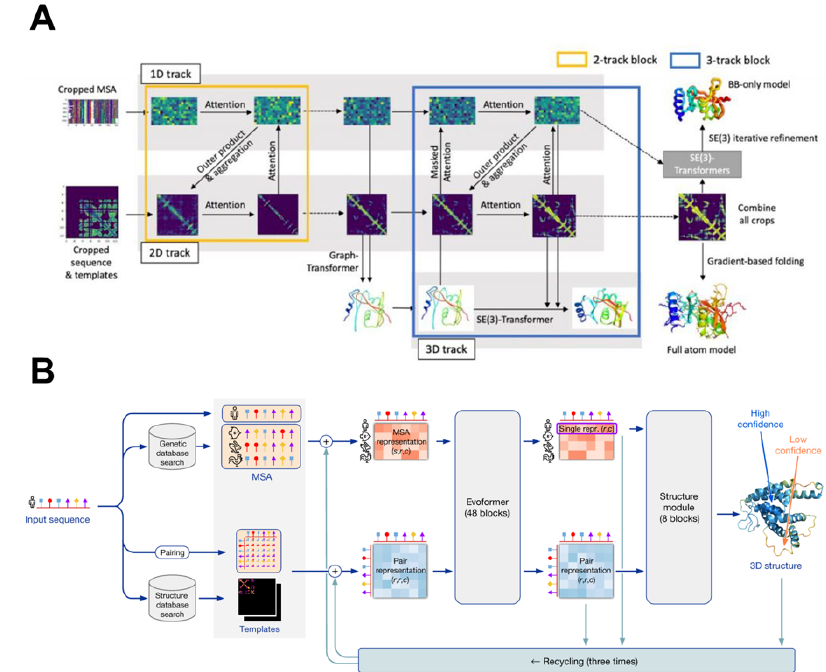
RoseTTAFold (https://github.com/RosettaCommons/RoseTTAFold), phát triển bởi David Baker và cộng sự, được xem như một công cụ tiên phong trong dự đoán cấu trúc protein ab initio. RoseTTAFold được xây dựng với kiến trúc mạng neuron 3-track (three-tracked neural network), bao gồm kiến trúc mạng 2-track kết hợp với một track cấu trúc làm việc trên tọa độ khung sườn 3D (Figure 2A). Trong kiến trúc mạng này, thông tin sẽ được di chuyển giữa thông tin trình tự amino acid (1D), bản đồ khoảng cách (2D), và tọa độ cấu trúc (3D) để tìm kiếm và liên kết mối quan hệ giữa trình tự, khoảng cách, và tọa độ. Mô hình 3D của protein được xây dựng bằng cách kết hợp và trung bình hóa các thông tin 1D, khoảng cách 2D, và dự đoán hướng xoay; sau đó, mô hình cuối được tạo dựng thông qua hai hướng tiếp cận: pyRosetta và mô hình SE(3)-Transformer40.
AlphaFold 2 (https://github.com/google-deepmind/alphafold) là hệ thống trí tuệ nhân tạo phát triển bởi DeepMind được sử dụng trong dự đoán cấu trúc 3D của protein, và được xem là lời giải cho bài toán gấp cuộn protein 41. So sánh với RoseTTAFold, AlphaFold2 sử dụng kiến trúc ba module: Module đầu vào: AlphaFold2 tìm kiếm các trình tự tương đồng với trình tự truy vấn trong các cơ sở dữ liệu (UniRef90, UniClust30, BFD,…) và trích xuất các thông tin đồng tiến hóa (co-evolutionary) từ kết quả sắp gióng cột trình tự (multiple sequences alignment – MSA) cũng như ma trận khoảng cách cặp (pairwise distance matrix – PDM). Module Evoformer: Đây được xem như máy phiên dịch các thông tin có được từ module trước bao gồm kết quả MSA và PDM. Lợi ích quan trọng nhất của module này chính là việc nó có thể chuyển đổi thông tin qua lại giữa biểu diễn MSA và PDM; thông tin MSA có thể được giải thích dưới dạng thông tin PDM và ngược lại; từ đó, các thông tin có thể được cải thiện lẫn nhau và đem lại kết quả dự đoán tốt hơn. Module cấu trúc: bao gồm 8 block, module này thu nhận các thông tin có được từ module Evoformer cùng với khung sườn protein để dự đoán cấu trúc 3D của protein. Ngoài ra, AlphaFold2 còn sử dụng thêm cơ chế tái chế (recycling) để tinh chỉnh cấu trúc, tăng độ chính xác của mô hình (Figure 2B). Chính vì vậy, cấu trúc dự đoán từ AlphaFold2 cho ra kết quả có độ tin cậy cực kì cao. So sánh giữa kết quả xác định cấu trúc bằng các phương pháp thực nghiệm và cấu trúc dự đoán cho thấy độ lệch RMSD rất thấp, chỉ từ 1-2 Å (Figure 3).
ESMFold (https://esmatlas.com/resources?action=fold) được phát triển bởi Meta-AI và dựa trên mô hình ngôn ngữ lớn (Large Language Model). ESMFold dựa vào thông tin biểu diễn từ mô hình ngôn ngữ để trực tiếp dự đoán cấu trúc cho trình tự đầu vào, thay vì sử dụng thông tin MSA như AlphaFold2 hay RoseTTAFold. Sự khác biệt về kiến trúc kể trên giúp tiết kiệm tài nguyên CPU dùng cho MSA và giảm số chiều dữ liệu, giúp ESMFold dự đoán nhanh hơn AlphaFold2 từ vài lần đến vài chục lần 42.

Kiến trúc mô hình dự đoán cấu trúc protein của RoseTTAFold (A) và AlphaFold2 (B)

So sánh độ tương đồng giữa cấu trúc thực nghiệm và cấu trúc dự đoán từ AlphaFold2
ĐÁNH GIÁ VÀ TINH CHỈNH MÔ HÌNH DỰ ĐOÁN
Độ chính xác của mô hình dự đoán là một yếu tố quan trọng cho những nghiên cứu cơ chế phức tạp như thiết kế thuốc, khảo sát gắn protein (protein docking), và dự đoán chức năng protein. Ứng với từng phương pháp xác định hay dự đoán mà cấu trúc ba chiều protein có thể được chấp nhận ứng dụng cho từng nghiên cứu khác nhau (Table 1).Tuy nhiên, các khiếm khuyết vẫn có thể xuất hiện, đặc biệt với các cấu trúc dự đoán từ phương pháp không dựa trên khuôn 43.
Biểu đồ Ramachandran biểu diễn các góc quay ψ (psi) và φ (phi) của chuỗi bên protein được sử dụng để đánh giá chất lượng hóa học lập thể của protein. Mỗi tổ hợp quay phi-psi sẽ thuộc một vùng cấu trúc bậc hai nhất định, nếu cấu trúc protein dự đoán là đáng tin cậy thì hầu hết các cặp góc sẽ rơi vào những vùng cho phép trên biểu đồ 44.
SAVES (https://saves.mbi.ucla.edu/) là một gói đánh giá chất lượng của mô hình dự đoán được tích hợp nhiều công cụ khác nhau như VERIFY3D, ERRAT, và PROCHECK45, 46.
Độ chính xác và tiềm năng ứng dụng của cấu trúc protein xác định theo phương pháp thực nghiệm, mô hình hoá tương đồng, xâu chuỗi và dự đoán
|
Độ tương đồng giữa cấu trúc tham chiếu và mục tiêu |
Độ phân giải và độ chính xác của mô hình dự đoán |
Phương pháp |
Ứng dụng của mô hình dự đoán |
|
100% |
< 1 Å (100%) |
Tinh thể hóa tia X Cộng hưởng từ hạt nhân (NMR) |
Nghiên cứu cơ chế xúc tác |
|
Thiết kế và cải thiện phối tử | |||
|
50% |
1.5 Å (95%) |
Mô hình hóa tương đồng |
Dự đoán phối tử/thụ thể |
|
Xác định epitope kháng thể | |||
|
30% |
3.5 Å (80%) |
Xâu chuỗi |
Nghiên cứu tác động của đột biến điểm |
|
Cải thiện chất lượng cho cấu trúc NMR | |||
|
Không đáng kể |
4-8 Å (~80 amino acid được dự đoán chính xác) |
Dự đoán ab initio |
Kết hợp với bản đồ điện tử có độ phân giải thấp |
|
Nhận diện vùng bảo tồn của các amino acid trên bề mặt protein |
Tinh chỉnh cấu trúc (structure refinement) là bước quan trọng trong quá trình dự đoán để mô hình tiệm cận độ chính xác với cấu trúc thực tế thông qua quá trình điều chỉnh cấu trúc bậc hai và tái đóng gói chuỗi bên 47. Quá trình tinh chỉnh bao gồm hai bước chính: lấy mẫu và chấm điểm tương tự như mô phỏng cấu trúc.
ModRefiner (https://zhanggroup.org/ModRefiner/) cho phép tinh chỉnh protein với độ phân giải cao. Quá trình tinh chỉnh bắt đầu từ Cα đến mô hình hóa mạch chính và toàn bộ nguyên tử. Cả chuỗi bên và mạch chính đều linh động trong mô phỏng tinh chỉnh cấu trúc, trong đó việc tìm kiếm cấu trúc được định hướng bằng trường lực48.
MỘT SỐ ỨNG DỤNG MÔ HÌNH HÓA CẤU TRÚC
Đại dịch Covid-19 đã mở ra nhiều chiến lược nghiên cứu nhằm ức chế, điều trị, và phòng ngừa sự xâm nhiễm của SARS-CoV-2. Các nhà khoa học đã tìm kiếm và đánh giá khả năng ức chế các protein quan trọng trong việc nhân lên của virus như 3Clpro, Mpro, nsp12, nsp15 và nsp16. Kết quả cho thấy, nhiều hợp chất tổng hợp như saquinavir, aclarubicin, ZG-7 hay từ tự nhiên như absinthin, curcumin đều có tiềm năng ức chế cao với những protein nêu trên 49, 50, 51, 52.
Trong nghiên cứu năm 2020, Kar và cộng sự đã ứng dụng phương pháp mô hình hóa cấu trúc protein để thiết kế và đánh giá vaccine đa epitope ngừa bệnh COVID-19 do virus SARS-CoV-2. Sau khi dự đoán các epitope tiềm năng, nhóm tác giả đã sử dụng webserver trRosetta để dự đoán cấu trúc vaccine và sau đó nghiên cứu tương tác của vaccine với các thụ thể của hệ miễn dịch (Figure 4) 53. Tương tự trên nghiên cứu của Ariz và cộng sự nhằm thiết kế một loại vaccine cho bệnh đậu mùa khỉ, nhóm tác giả đã sử dụng tổ hợp các phương pháp tin sinh học cấu trúc, bao gồm mô hình hóa cấu trúc protein bằng I-TASSER và RoseTTAFold để mô hình hóa cấu trúc của vaccine54.

Kết quả mô phỏng cấu trúc vaccine đa epitope phòng SARS-CoV-2 của Kar và cộng sự. A) Thiết kế vaccine từ các epitope liên tục từ CTL, HTL và IFNγ; B) mô hình 3D của vaccine; C) đánh giá chất lượng cấu trúc thông qua ERRAT; D) đánh giá chất lượng cấu trúc bằng ProSA; E) đánh giá chất lượng lập thể cấu trúc bằng biểu đồ Ramachandran
Việc thiết kế kháng thể trung hòa trong bối cảnh đại dịch cũng là một chiến lược bên cạnh công cuộc phát triển vaccine. Bằng việc sử dụng phương pháp mô hình hóa tương đồng và machine learning, sử dụng cấu trúc protein gai của SARS-CoV-1 và hệ thống AS2TS, năm 2020, Thomas Desautels và cộng sự đã cho ra 20 mô hình kháng thể trung hòa cho SARS-CoV-2, có khả năng liên kết tốt với thụ thể ACE2 từ 89.263 mô hình55.
KẾT LUẬN
Mối quan tâm về quá trình gấp cuộn và tương tác protein-protein được xem như là vấn đề cốt lõi trong lĩnh vực sinh học cấu trúc. Dự đoán cấu trúc protein bằng điện toán đám mây đã ra đời như một chìa khóa mở ra cơ hội nghiên cứu sâu và rộng hơn cho giới khoa học cũng như rút ngắn chênh lệch giữa số lượng trình tự và cấu trúc xác định. Những thuật toán, hàm số vật lý và công nghệ dữ liệu mới ngày càng phát triển đã góp phần đẩy mạnh công cuộc giải mã cấu trúc protein. Năm 2021, AlphaFold 2 ra đời và được xem như là lời giải cho bài toán gấp cuộn protein đã tồn tại gần 50 năm. Những hiểu biết về cấu trúc đem lại cho các nhà nghiên cứu cái nhìn sâu hơn về cơ chế hoạt động của những đại phân tử sinh học này và ứng dụng cho việc phát triển vaccine, kháng thể, và thiết kế thuốc trong tương lai.
DANH MỤC TỪ VIẾT TẮT
MR : Nuclear Magnetic Resonance
EM: Electron Microscope
CSDL: Cơ Sở Dữ Liệu
PDB: Protein Data Bank
BLAST: Basic Local Alignment Search Tool
PSI-BLAST: Position-Specific Iterative Basic Local Alignment Search Tool
FPPS: Families of Structurally Similar Proteins
SCOP: Structural Classification of Proteins
DOPE: Discrete Optimized Protein Energy
MC: Monte Carlo
REM: Replica Exchange Monte Carlo
MCM: Monte Carlo-minimization
MDs: Molecular Dynamics Simulation
aMD: accelerated Molecular Dynamics
XUNG ĐỘT LỢI ÍCH
Các tác giả cam kết không có xung đột lợi ích.
ĐÓNG GÓP CỦA CÁC TÁC GIẢ
Lê Mạnh Liêm: viết, tổng hợp và chỉnh sửa bản thảo
Nguyễn Văn Minh Thường: viết, tổng hợp và chỉnh sửa bản thảo
Đinh Thuận Thiên: lên ý tưởng, viết, tổng hợp và chỉnh sửa bản thảo
Trần Văn Hiếu: lên ý tưởng, tham gia chỉnh sửa bản thảo và chấp thuận bản thảo.
Tất cả các tác giả đồng ý với bản thảo cuối cùng.